Edge Energy & 吸收边
(16 min 37 s reading) 什么是 E0?怎么选 E0?
视频:E0, Edge Energy & 吸收边 | XAFS数据处理参数讨论 Bilibili, YouTube
阅读提示1:参考阅读有 X‐Ray Spectrom., 37: 572-584., 《XAFS for Everyone》 和 《Introduction to XAFS: A Practical Guide to X-ray Absorption Fine Structure Spectroscopy》。
阅读提示2:该文只用于参考和建议,非标准。各个课题遭遇的数据差异巨大,我的建议不一定适合你的数据处理,请谨慎使用。
阅读提示3:回答仅代表我一个人的看法,不代表正确解决问题的办法。我有不知道答案的问题,也有不懂回答错误的问题。如果我有讲错地方,请告诉我还有其他人。
如果来不及看完全文
- E0 是指让电子离开原子的能量。这个定义模棱两可,看了等于没看。
- 在 Athena 中,E0 通常是可以自己人为来定义,没有固定套路。例如以下几类:
a. 吸收先谱图跳起来的点;
b. XANES 一阶微分(first derivative) 中的第一个顶点;若有 pre-edge 存在,建议选择强度最高的顶点 (我个人最为推荐);
c. XANES 中起跳后第一个峰 (1st peak in white line region)。
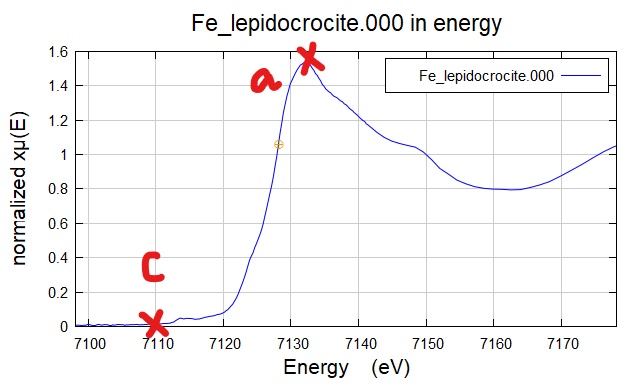
Figure 1. 选择E0 的 a 点(一开始出现吸收)和 c 点(吸收最高)。
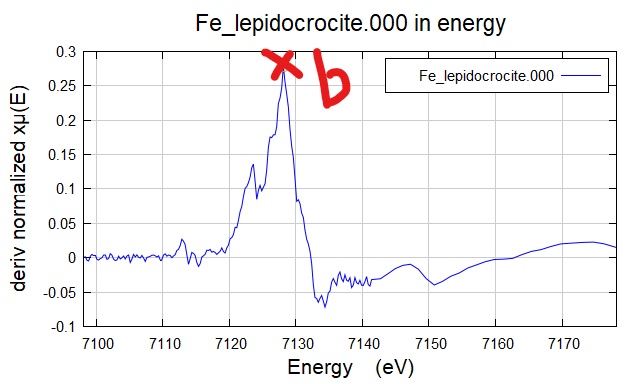
Figure 2. 选择E0 的 b 点(一阶微分中最高)。
- 严格来说,E0 不可以作为判断氧化态相对高低的唯一标准。
- 在报告 edge energy 的时候,例如写文章,仍用 Athena 中选择的 E0 数值。
- 在 Athena 中使用任一标准理论上都可以,但要注意:
a. 在后续 Artemis 拟合时,|E0| ≤ 10 eV;
b. 在写文章的时候需要明确指出自己选择的标准;
c. 在需要相互比较的 XAFS 数据时,尽可能使用同一标准。 - 在 Artemis 中,E0 是对在 Athena 中的 E0 进行修正。这仅仅是出于对 EXAFS 拟合目的,不是对 E0 本质上的修改。
- EXAFS 拟合中通常对不同的路径 (path) 只会有一个相同 E0 变量,因为电子都是从同一个中心原子逃逸。除非存在样品中是两种及两种以上不同氧化态的混合物,对各自组分定义不同的 E0。实际情况中需要考虑变量数量上限 (number of independent points), 很难有给 E0 多个变量的情况。
进一步讨论
1. 理论定义
E0 的定义是来源于光电子相关公式的定义:
注:me 电子质量, ħ 狄拉克常数。
我们关心的是入射光子能量(E)和波数(k,也就是 EXAFS 中 k 空间的 x 轴) 之间的关系。在入射光子能量 E 中,包含了两个部分:让内层电子逃离原子的能量和逃离之后剩余能量变为动能。而波数 k 只和动量相关,所以需要移除“让电子离开原子的”能量,也就是 E0 。
Edge 的定义是:
The energy at which there is a sharp rise (discontinuity) in the (linear) absorption coefficient of X-rays by an element. (IUCr 2011.)
同样是一个说了等于没说的定义。因为这一个 "sharp rise",从开始吸收能量到达到最高值,是发生在了一段能量距离,而不是精准的一个点。 很多时候我们也用“edge”来描述一个某个元素做的某个边。比如 Fe K-edge EXAFS 也就是铁元素的 K 边,那就是说测量的是 Fe 元素在 7,112 eV 附近的同步辐射精细衍射吸收谱(-150 ~ +1000 eV)。
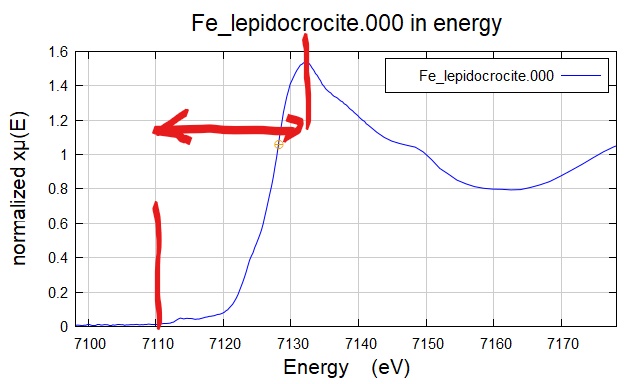
Figure 3. 纤铁矿的 Fe K-edge XANES 有从大约 7,110 ~ 7,733 eV 都可以看作 edge。
给出 E0 和 Edge 的定义后,我们在实际操作中常常把它们两个画上等号。
2. E0 出现的地方
我们需要把上述的理论定义具象化,在 XAFS 中常提及的 E0 出现在:
1. 对参考金属单质 (reference spectra) 进行校正 (calibration) 的时候。例如:利用 Fe 金属来对含铁样品进行能量校正,那么 Fe 金属的 K-edge E0 在 7,112 eV。
2. 在 Athena 中对样品选择在 XANES 一阶微分中的最高峰作为这个样品的 E0。例如:对 Fe_lepidocrocite 样品(纤铁矿),选择了 7128.2 eV 作为 它的 E0。
3. 在 Artemis 拟合中,会用到 E0 来帮助 EXAFS 微调,使拟合结果更加吻合实验数据。
既然它们出现在了不同的地方,跟他人交流 "E0" 时有必要先讲明是什么 E0,否则很容易出现误会。
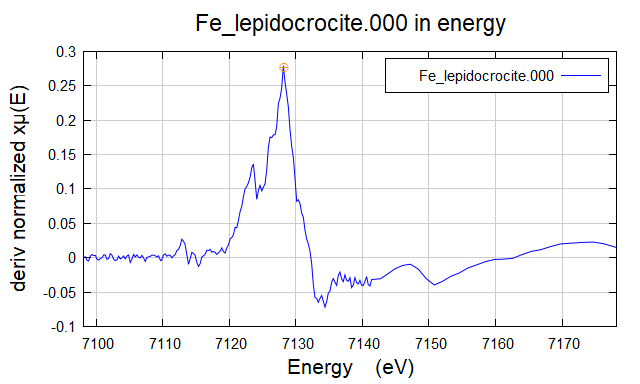
Figure 4. 在 XANES 一阶微分中选择了最高点(黄色小圆点)。
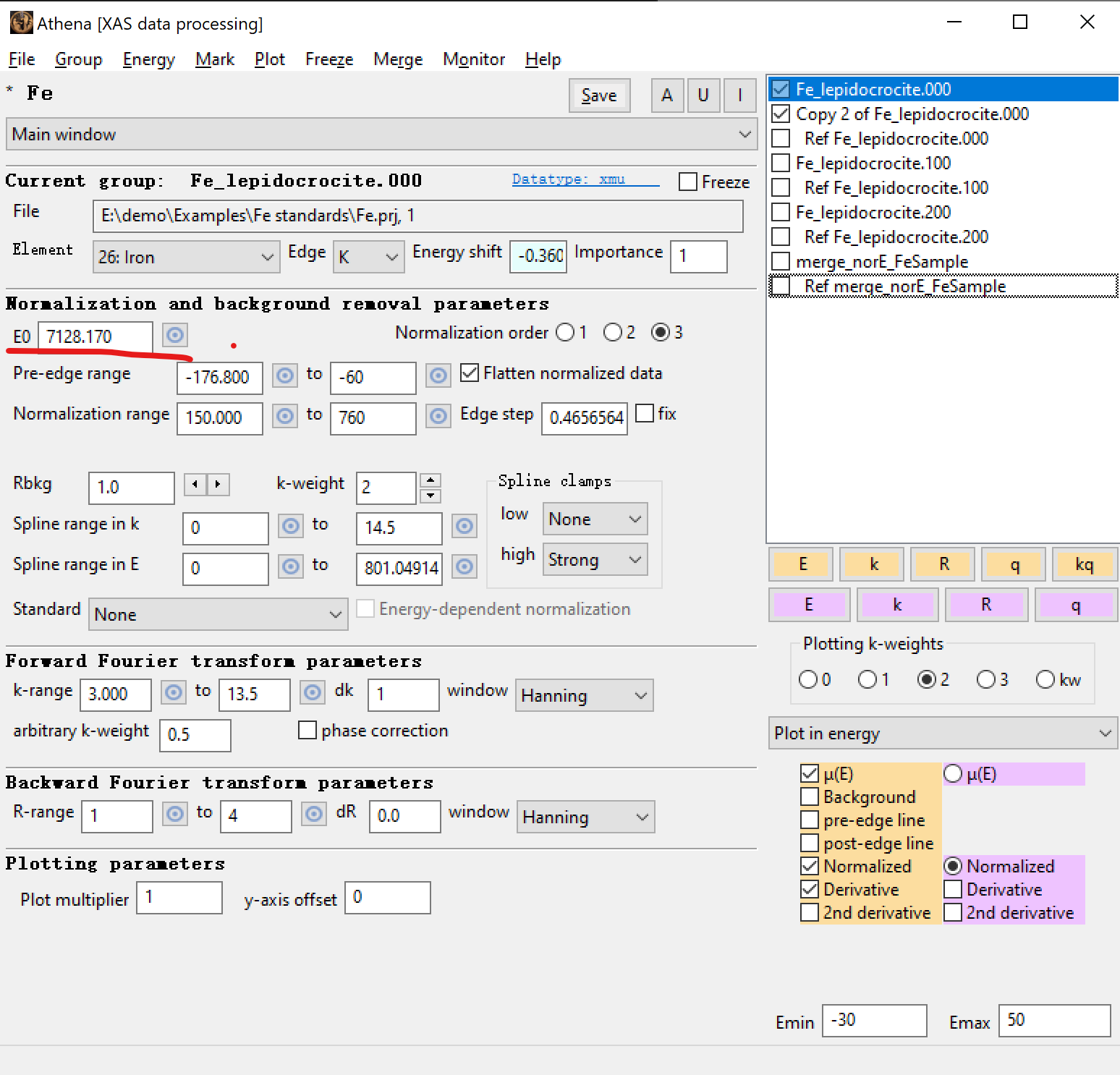
Figure 5. 在 Athena 界面中显示 7128.2 eV(对应 Figure 4 中的小黄点)。
3. “选择” E0
单质(常用于校正目的)
在一阶微分中选择第一个峰,具体能量数值可以参考 X-ray Absorption Edges。然而最近在 Ifeffit 讨论中有提及到 W L2 和 L3-edge 的能量不是 X-ray Absorption Edges 标出的 11,544 和 10,207 eV。但不必太过惊慌,目前大部分人依然参考这些能量值作为标准,建议在发表文章的时候讲明用于校正的能量值即可。
样品(已知标准样品或未知样品)
在 Athena,可以参考的标准有很多(不是以推荐顺序来列的):
1. 一阶微分中的第一个峰 (first inflection point);
2. 一阶微分中最高的峰 (largest peak in 1st derivative XANES);
3. 吸收最强区间 (white line) 的第一个峰,常常是吸收能量起跳后第一个峰;
4. (在归一化后)取 y = 0.5 时的能量数值 (halfway up the edge);
5. 用其他(类似)样品得到的 E0 来定义;
6. 在吸收发生开始的能量数值(onset of the edge)。
……
不难想象,不同的定义会给出不同的 E0 的数值。没有必要对于同一个化合物给出的不同E0 值感到疑惑。正确做法是去在文中寻找作者是如何定义 E0。在《XAFS for Everyone》 Box 10.4 中写到:
But the bottom line is that it (E0) WORKS! In the EXAFS region, the spectrum behaves as if there are electrons with a well-defined momentum, meaning E0 is well defined too.
在《Introduction to XAFS: A Practical Guide to X-ray Absorption Fine Structure Spectroscopy》中作者也说道:
All of these metrics (E0) are sufficient because we just need an initial guess; a few electron volts error doesn’t matter, because the energy zero is fine-tuned in later analysis (meaning Artemis).
说大白话就是:E0 能使就行。比如让你老板觉得行,比如让你的 Artemis 里面拟合出来的 E0 绝对值小于等于 10 eV。
EXAFS中拟合 E0
在视频《从 EXAFS 方程讨论拟合变量 | Artemis | 同步辐射吸收谱(XAS)数据处理》(Bilibili, YouTube) 中讨论过 E0 会影响 k 空间开始的地方 (k = 0),从而影响相位 (phase)。在EXAFS中拟合的具体体现就是会影响原子距离 (R)。
Bruce Ravel 的讲义里面提到在 Artemis 中限制变量数量可以优先考虑 S02 和 E0,即对于同一个化合物或是样品,它们在每一条 path 中都假定是一样的:
S02: This is a parameter of the central atom and has somthing to do with the relaxation of electrons around the core-hole. In copper metal (one of his presentation examples), S02 is the same for all paths.
E0: In a single data set, single FEFF calculation fit, this parameter is used to align the wavenumber grids of the data and theory. In copper metal (one of this presentation examples), E0 is the same for all paths.
当有人问为什么谁谁谁的文献一个标准样品每一个 path 都有一个不同的 E0 的时候,大约只会是两种答案:1. 他/她用的不是 Artemis 来拟合;2. 他/她是菜狗。
E0 只出现在 Artemis 里面,目的只用来提高拟合结果,没有实际的物理意义。所以在汇报 E0 的时候,仍选择在 Athena 里面的数值,例如 Figure ,5. 中的 7,128.2 eV。
Pre-edge (讨论以 K-edge 为主)
Pre-edge常见于过渡金属XANES中,顾名思义发生在 "edge" 之前,在主要的跃迁中间有较小的峰。因为其形状不一,pre-edge 经常用作于指纹信息 (fingerprint information),可以快速定性地判定物种信息。这是因为 pre-edge 的形成条件与 d 轨道结构紧密相关,而 d 轨道上通常又承载了价电子 (valence electron)、配位数 (coordination number)和几何结构(四面体、八面体等)之类的结构信息。
针对一个具有某种几何结构的某种元素化合物,在某种价态上会有pre-edge 特征峰。如果拿未知样品和已知样品的 XANES 特征(包含 edge 和 pre-edge)进行比较,可以大致得到一个定性的结论。
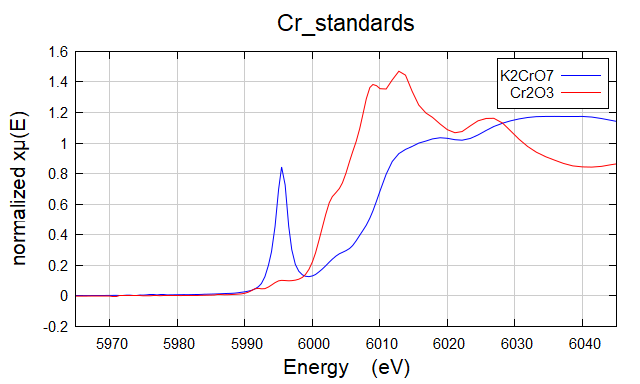
Figure 6. Cr K-edge XANES for Cr2O3 (+3, octahedral CrO6 geometry) and K2Cr2O7 (+6, tetrahedral CrO4 geometry).
主要跃迁 (edge) 形成:光谱选择规则允许
l= ± 1:主要跃迁
Pre-edge 的形成原因与原子能级之间的跃迁存在选择定则 (selection rule) 有关。简单来说,光谱跃迁需要满足 l= ± 1 和 j = 0, ± 1 两个标准,尤其是前者。
这个要求是要求两个轨道的量子数相差为1。量子数也就是原子轨道:s, p, d, f, g...对应的量子数是 0, 1, 2, 3, 4, 5... 按照这样的规则,如果是在 s 轨道 (l = 0) 的电子只能去 p 轨道 (l = 1);如果是 p 轨道的电子则可以去 s 或者 d 轨道 (l = 2)。
这一个规则是决定了主要吸收,也就是XANES中的 edge。对 K-edge,是 1s 电子跃迁到有空余位置的 np 轨道,比如 Fe K-edge (1s → 4p)、Mo K-edge (1s → 5p)。另一方面按照这样的规则 s 轨道电子到 d 轨道是不允许的。
部分跃迁 (pre-edge) 形成:对称性允许和 p-d “杂化”
根据分子轨道理论 (molecular orbital theory),分子轨道是由原子轨道线性组合得到。如果发生跃迁,需要满足对称性一致的要求。为了成键,主要考虑 s, p, d 轨道。其中 s 轨道的对称性最高(球形),可以自由满足跟其他轨道的条件,不用考虑对称性限制。剩下的就是考虑 p 和 d 轨道。 为了方便解释这个问题,我们可以借用“杂化”的概念来进一步描述 Td, Oh 和 D4h 对称的分子轨道结构。
具有 Td 对称性的分子为对称四面体(参考 CH4)。px,y,z 和 dxy,xz,yz 属于 T2 不可约对称性。因为对称性匹配,p 和 d 轨道发生“杂化”,形成分子轨道。1s 电子在这样的条件下可以发生 electric quadrupole transition,跃迁到 p-d “杂化”轨道上。核心仍然是 1s 电子跃迁到 p 轨道上,但由于与 d 轨道“杂化”(通常是 (n-1) d 轨道),能量比”未杂化“轨道高,pre-edge 会出现在 edge 之前。
具有 Oh 对称性的分子为对称八面体(参考 SF6)。px,y,z 和 dxy,xz,yz 没有共同的对称性所以无法形成“杂化”,也就是说完全对称的六配位化合物没有 pre-edge。
具有 D4h 对称性的分子通常是平面四配位(参考 [Ni(CN)4]2-)。px,y,z 和 dxy,xz,yz 同属于 Eg 不可约对称性。所以同样也允许 1s 电子跃迁到 p-d “杂化”轨道,也会有 pre-edge。
在上述介绍中,特地强调了是“完全对称”配位构型。这是因为在变形八面体 (distorted octahedral) 中,p-d “杂化”依然可以发生,出现很小的 pre-edge。所以六配位 ≠ no pre-edge。对于其他构型也是如此:如果几何结构存在变形 (distortion),pre-edge 会受到影响。
对称性不是唯一的影响因素,d 轨道电子数量同样可以影响 pre-edge。如果 3d 轨道在发生跃迁前已经部分被价层电子占满,即使有 p-d “杂化”轨道,1s 电子的跃迁会受到价电子的影响。内层电子被激发前价电子越多的元素,1s 电子到 p-d “杂化”轨道的概率越小,pre-edge 的强度越小。 半充满的电子结构同样会受到影响,例如 Fe3+ (3d5) 的XANES只有很小的 pre-edge,但是 Fe6+ (3d0) 有明显 pre-edge。
由于 pre-edge 的变化和形状实在太大且复杂,不会在这篇文章里面展开讲了。如果需要了解,从同一元素开始(比如自己手头上关心的实验数据)。Pre-edge 一大便利是可以直接观察,但常常会出现“比较得不够仔细”或者“比较错了对象”。如果要保证严格的论述,建议留心如下细节:
a. 判断未知样品的几何结构还有价态;
b.在做实验前,阅读文献了解相同元素的 XANES 是如何随着几何结构和价态改变的,观察标准样品的晶体结构确认一致性;
c. 根据上述两步得到的信息,准备标准样品并跟随未知样品一起去同步辐射实验室测试;
d. 得到数据后,先确认标准样品的 XANES 是否和文献中一致,再去跟未知样品比较,甚至是可以做 Linear combination fittings。
(对于像 Fe 这样烂大街的元素,XANES 分析一抓一大把。但是像 Ga 比较冷门的元素,做破头都不知道在干嘛)
还有更多具体的跨元素例子和讨论,可以去参考一开始列出来的文献。
感谢你的阅读。